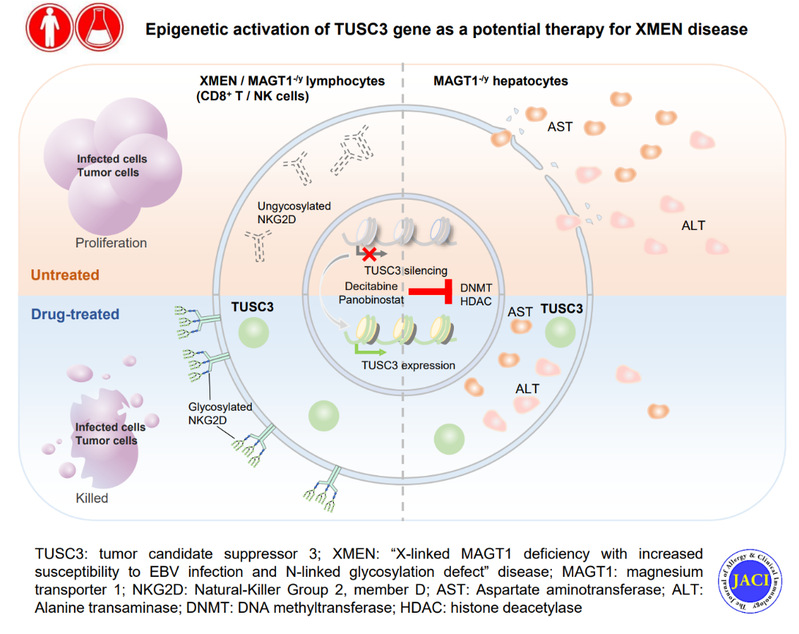
“X-连锁MAGT1缺陷伴随EBV易感性增加及N-连接糖基化缺陷”(XMEN)疾病是一种罕见的联合免疫缺陷疾病(CID),由镁离子转运蛋白1 (MAGT1)基因发生功能丧失性突变所引起。MAGT1的功能缺陷会损害镁离子转运及系列蛋白的N-糖基化,从而导致多个关键免疫受体(如NKG2D)表达缺失,继而诱发免疫系统异常、慢性EBV感染和肿瘤形成。此外,非病毒性肝酶异常升高也是XMEN患者的主要病征。目前尚无针对XMEN疾病的特定有效治疗方法。XMEN患者存在反复感染,生存质量较差,大部分患者20-40岁死亡。

2023年4月21日,复旦大学生物医学研究院/附属儿科医院罗敏/卢智刚团队和复旦大学附属儿科医院侯佳合作,在Journal of Allergy and Clinical Immunology在线发表了Epigenetic activation of the TUSC3 gene as a potential therapy for XMEN disease论文。
MAGT1和TUSC3在镁离子转运和蛋白N-糖基化功能上表现出功能冗余。该研究首先利用组织样本及多个数据库分析了MAGT1和TUSC3的人体组织表达谱,并系统分析其与XMEN患者病征的相关性。结果显示TUSC3虽然在多种器官组织中广谱表达,但是在免疫系统和肝脏中并不表达,与XMEN患者的主要病变组织一致;也提示重新激活TUSC3的表达有可能恢复XMEN细胞的功能。
目前XMEN疾病相关的研究模型非常有限。XMEN是一种极罕见的遗传疾病,患者来源的细胞极其稀少、细胞增殖数量有限;而小鼠模型与人的表型存在一定差异。该研究利用人来源的NKL细胞构建了MAGT1 KO细胞系,成功模拟了XMEN患者来源淋巴细胞的缺陷表型,为研究XMEN疾病的发病机制和治疗策略提供了可靠的细胞模型平台。
基于以上细胞模型,作者证明TUSC3能够显著恢复MAGT1 KO 所导致的免疫缺陷。研究继而利用该模型进行小分子药物筛选,鉴定出两种表观遗传药物:地西他滨和帕比司他,能够显著上调 TUSC3表达,并修复XMEN免疫缺陷和肝酶异常;药物的治疗效果在XMEN患者来源的免疫细胞上得到了进一步的证明。该研究为XMEN疾病的有效治疗提供了新的策略。
复旦大学附属儿科医院硕士丁浩东为该文的第一作者;复旦大学生物医学研究院/附属儿科医院罗敏研究员、生物医学研究院卢智刚研究员,以及复旦大学附属儿科医院临床免疫科侯佳副主任医师为该论文的共同通讯作者。
原文链接:https://doi.org/10.1016/j.jaci.2023.04.003